![[advertisement] concertmedical](../../files/bk-nysora-ad.jpg)
Local Anesthetics: Clinical Pharmacology and Rational Selection
|
Author: Jeff Gadsden Local anesthetics (LAs) prevent or relieve pain by interrupt- ing nerve conduction. They bind to specific receptor sites on the sodium (Na+) channels in nerves and block the movement of ions through these pores. Both the chemical and pharmacologic properties of individual LA drugs determine their clinical properties. This chapter discusses the basics of the mechanism of action of LAs, their clinical use, and systemic toxicity prevention and treatment. Nerve Conduction Nerve conduction involves the propagation of an electrical signal generated by the rapid movement of small amounts of several ions (Na+ and potassium K+) across a nerve cell membrane. The ionic gradient for Na+ (high extracellularly and low intracellularly) and K+ (high intracellularly and low extracellularly) is maintained by a Na+-K+ pump mechanism within the nerve. In the resting state, the nerve membrane is more permeable to K+ ions than to Na+ ions, resulting in the continuous leakage of K+ ions out of the interior of the nerve cell. This leakage of cations, in turn, creates a negatively charged interior relative to the exterior, resulting in an electric potential of 60-70 mV across the nerve membrane. Receptors at the distal ends of sensory nerves serve as sensors and transducers of various mechanical, chemical, or thermal stimuli. Such stimuli are converted into minuscule electric currents. For example, chemical mediators released with a surgical incision react with these receptors and generate small electric currents. As a result, the electric potential across a nerve membrane near the receptor is altered, making it less negative. If the threshold potential is achieved, an action potential results, with a sudden increase in the permeability of the nerve membrane to Na+ ions and a resultant rapid influx of positively charged Na+ ions. This causes a transient reversal of charge, or depolarization. Depolarization generates a current that sequentially depolarizes the adjacent segment of the nerve, thus "activating" the nerve and sending a wave of sequential polarization down the nerve membrane. Repolarization takes place when sodium permeability decreases and K+ permeability increases, resulting in an efflux of K+ from within the cell and restoration of the electrical balance. Subsequently, both ions are restored to their initial intracellular and extracellular concentrations by the Na+-K+-adenosine triphosphate pump mechanism. Because the rapid influx of Na+ ions occurs in response to a change in the transmembrane potential, Na+ channels in the nerve are characterized as "voltage gated." These channels are protein structures with three subunits that penetrate the full depth of the membrane bilayer and are in communication with both the extracellular surface of the nerve membrane and the axoplasm (interior) of the nerve. LAs prevent the generation and conduction of nerve impulses by binding to the α subunit of the Na+ channel and preventing the influx of Na+ into the cell, halting the transmission of the advancing wave of depolarization down the length of the nerve. A resting nerve is less sensitive to a LA than a nerve that is repeatedly stimulated. A higher frequency of stimulation and a more positive membrane potential cause a greater degree of transmission block. These frequency- and voltage- dependent effects of LAs occur because repeated depolarization increases the chance that a LA molecule will encounter a Na+ channel that is in the activated, or open, form-as opposed to the resting form-which has a much greater affinity for LA. In general, the rate of dissociation from the receptor site in the pore of the Na+ channel is critical for the frequency dependence of LA action. Structure-Activity Relationship of Local Anesthetics The typical structure of a LA consists of hydrophilic and hydrophobic domains separated by an intermediate ester or amide linkage. The hydrophilic group is usually a tertiary amine, and the hydrophobic domain is an aromatic moiety. The nature of the linking group determines the pharmacologic properties of LA agents. The physicochemical properties of these agents largely influence their potency and duration of action. For instance, greater lipid solubility increases both the potency and duration of their action. This is due to a greater affinity of the drug to lipid membranes and therefore greater proximity to its sites of action. The longer the drug remains in the vicinity of the membrane, rather than being replaced by the blood, the more likely the drug will be to effect its action on the Na+ channel in the membrane. Unfortunately, greater lipid solubility also increases toxicity, decreasing the therapeutic index for more hydrophobic drugs.
The pKa (the pH at which 50% of the drug is ionized and 50% is present as base) of the LA is related to pH and the concentrations of the cationic and base forms by the Henderson-Hasselbalch equation: pH = pKa + log ([base]/ [cation]). The pKa generally correlates with the speed of onset of action of most amide LA drugs; the closer the pKa to the body pH, the faster the onset. The coexistence of the two forms of the drug - the charged cation and the uncharged base-is important because drug penetration of the nerve membrane by the LA requires the base (unionized) form to pass through the nerve lipid membrane; once in the axoplasm of the nerve, the base form can accept a hydrogen ion and equilibrate into the cationic form. The cationic form is predominant and produces a blockade of the Na+ channel. The amount of base form that can be in solution is limited by its aqueous solubility. An ester or an amide linkage is present between the lipophilic end (benzene ring) and the hydrophilic end (amino group) of the molecule. The type of linkage determines the site of metabolic degradation of the drug. Ester-linked LAs are metabolized in plasma by pseudocholinesterase, whereas amide-linked drugs undergo metabolism in the liver. The Onset and Duration of Blockade Local Anesthetic Diffusion A mixed peripheral nerve or nerve trunk consists of individual nerves surrounded by an investing epineurium. When a LA is deposited in proximity to a peripheral nerve, it diffuses from the outer surface toward the core along a concentration gradient. Consequently, nerves located in the outer mantle of the mixed nerve are blocked first. These fibers are usually distributed to more proximal anatomic structures than those situated near the core of the mixed nerve and often are motor fibers. When the volume and concentration of LA solution deposited in the vicinity of the nerve are adequate, the LA eventually diffuses inward to block the more centrally located fibers. In this way, the block evolves from proximal structures to distal structures. Smaller amounts and concentrations of a drug only block the nerves in the mantle and smaller and more sensitive central fibers. Onset of Blockade In general, LAs are deposited as close to the nerve as possible, preferably into the tissue sheaths (e.g., brachial plexus, lumbar plexus) or epineurial sheaths of the nerves (e.g., femoral, sciatic). The actual site of local anesthetic injection and its relationship to the nerve structures is much better understood since the advent of the use of ultrasound guidance during nerve blockade. Intraneural or sub-epineural injections reportedly occur relatively frequently with some peripheral nerve blocks. The available data indicate that such injections result in faster onset of blockade, most likely due to the intimate proximity of LA to the nerve tissue. This is hardly surprising because the LA must diffuse from the site of injection to the nerve, the site of action. However, intraneural injections should not be recommended as a safe practice despite limited reports suggesting that intraneural injections do not inevitably lead to nerve injury. These data must be interpreted with caution because the term intraneural injection is often used loosely to denote injections within epineurium or even tissue sheaths that envelop the peripheral nerves or plexi. However, neurologic injury is much more likely to occur should an intraneural injection occur intrafascicularly. The rate of diffusion across the nerve sheath is determined by the concentration of the drug, its degree of ionization (ionized LA diffuses more slowly), its hydrophobicity, and the physical characteristics of the tissue surrounding the nerve. Duration of Blockade The duration of nerve block anesthesia depends on the physical characteristics of the LA and the presence or absence of vasoconstrictors. The most important physical characteristic is lipid solubility. In general, LAs can be divided into three categories: short acting (e.g., 2-chloroprocaine, 45-90 minutes), intermediate duration (e.g., lidocaine, mepivacaine, 90-180 minutes), and long acting (e.g., bupivacaine, levobupivacaine, ropivacaine, 4-18 hours). The degree of block prolongation with the addition of a vasoconstrictor appears to be related to the intrinsic vasodilatory properties of the LA; the more intrinsic vasodilatory action the LA has, the more prolongation is achieved with addition of a vasoconstrictor. Although this discussion is in line with current clinical teaching, it is really more theoretical than of significant clinical relevance. For instance, dense blocks of the brachial plexus with 2-chloroprocaine are likely to outlast weak poor quality blocks with bupivacaine. In addition, classical teaching does not take into account the nerve to be anesthetized. As an example, a sciatic nerve block with bupivacaine lasts almost twice as long as an interscalene or lumbar plexus block with the same drug dose and concentration. These differences must be kept in mind to time and predict the resolution of blockade properly.
Differential Sensitivity of Nerve Fibers to Local Anesthetics Two general rules apply regarding susceptibility of nerve fibers to LAs: First, smaller nerve fibers are more susceptible to the action of LAs than large fibers (Figure 1). Smaller fibers are preferentially blocked because a shorter length of axon is required to be blocked to halt the conduction completely. Second, myelinated fibers are more easily blocked than nonmyelinated fibers because local anesthetic pools near the axonal membrane. This is why C-fibers, which have a small diameter (but are unmyelinated), are the most resistant fibers to LA. The sensitivity of a fiber to LAs is not determined by whether it is sensory or motor. In fact, muscle proprioceptive afferent (A-beta) and motor efferent fibers (A-alpha) are equally sensitive. These two types of fibers have the same diameter, which is larger than that of the A-gamma fibers that supply the muscle spindles. It is the more rapid blockade of these smaller A-gamma fibers, rather than of the sensory fibers, that leads to the preferential loss of muscle reflexes. Similarly, in large nerve trunks, motor fibers are often located in the outer portion of the bundle and are more accessible to LA. Thus motor fibers may be blocked before sensory fibers in large mixed nerves. The differential rate of blockade exhibited by fibers of varying sizes and firing rates is of considerable practical importance. Fortunately, the sensation of pain is usually the first modality to disappear; it is followed by the loss of sensations of cold, warmth, touch, deep pressure, and, finally, loss of motor function, although variation among patients and different nerves is considerable. Local Anesthetics and pH LA drugs, as previously described, pass through the nerve membrane in a nonionized lipid-soluble base form; when they are within the nerve axoplasm, they must equilibrate into an ionic form to be active within the Na+ channel. The rate-limiting step in this cascade is penetration of the LA through the nerve membrane. LAs are unprotonated amines and as such tend to be relatively insoluble (Figure 2). For this reason, they are manufactured as water-soluble salts, usually hydrochlorides. Although LAs are weak bases (typical pKa values range from 7.5-9), their hydrochloride salts are mildly acidic. This property increases the stability of LA esters and any accompanying vasoconstrictor substance. However, this means that the cationic form predominates in solution. 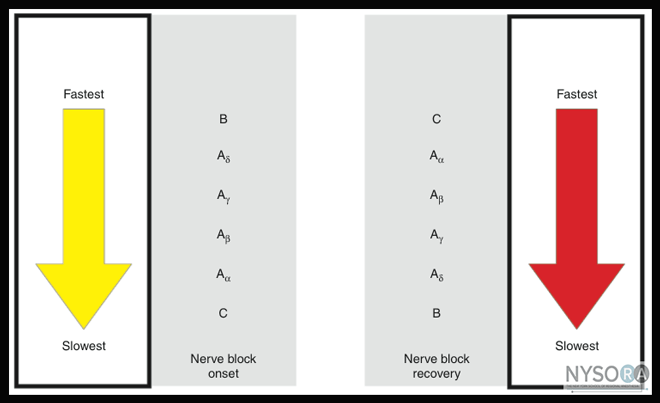
Figure 1: Differential rate of nerve blockade. For this reason, sodium bicarbonate (NaHCO3) is often added to LA. This increases the amount of drug in the base form, which slightly shortens the onset time. Obviously, the limiting factor for pH adjustment is the solubility of the base form of the drug. Unfortunately, only small changes in pH can be achieved by the addition of bicarbonate because of the limited solubility of the base. As such, only small decreases in onset time are realized. For instance, with the alkalinization of bupivacaine, an increase in the amount of base in solution is limited by the minimal solubility of free base in solution. For each LA, there is a pH at which the amount of base in solution is maximal (a saturated solution). Further increases in pH result in precipitation of the drug and do not produce an additional shortening of onset time.
Protein Binding LAs are in large part bound to plasma and tissue proteins. However, they are pharmacologically active only in the free, unbound state. The most important binding proteins for LAs in plasma are albumin and alpha1-acid glycoprotein (AAG). The binding to AAG is characterized as high-affinity but low-capacity binding; hence LAs bind to AAG preferentially compared with albumin. However, binding to AAG is easily saturated with clinically achieved blood levels of LA. Once AAG saturation occurs, any additional binding is to albumin. Albumin can bind LA drugs in plasma in concentrations many times greater than those clinically achieved. 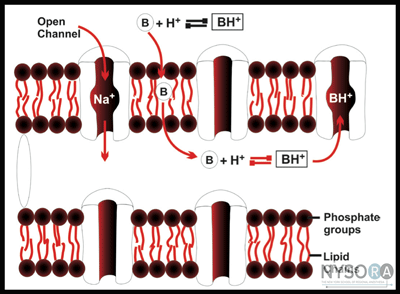 Figure 2: Local anesthetics and pH. Local anesthetics pass through the nerve membrane in a nonionized, lipid- soluble base form. When they are within the nerve axo- plasm, they must equilibrate into an ionic form to exert their action on the Na+ channel. Note that the fraction of drugs bound to protein in plasma correlates with the duration of LA activity: bupivacaine > etidocaine > ropivacaine > mepivacaine > lidocaine > procaine and 2-chloroprocaine. However, no direct relationship exists between LA plasma protein binding and binding to specific membrane-bound Na+ channels. Rather, there is a direct correlation between protein binding and lipid solubility, as there is for all drugs. The more lipid soluble the drug, the more likely it will remain in the lipid-rich environment of the axonal membrane where the Na+ channel resides. The degree of protein binding of a particular LA is concentration dependent and influenced by the pH of the plasma. The percentage of drug bound decreases as the pH decreases. This is important because with the development of acidosis, as may occur with LA-induced seizures or cardiac arrest, the amount of free drug increases. The magnitude of this phenomenon varies among LAs, and it is much more pronounced with bupivacaine than with lidocaine. For instance, as the binding decreases from 95% to 70% with acidosis, the amount of free bupivacaine increases from 5% to 30% (a factor of 6), although the total drug concentration remains unchanged. Because of this increase in free drug, acidosis renders bupivacaine markedly more toxic. Systemic Toxicity of Local Anesthetics In addition to interrupting peripheral nerve conduction, LAs interfere with the function of all organs in which the conduction or transmission of nerve impulses occurs. For instance, they have important effects on the central nervous system (CNS), the autonomic ganglia, the neuromuscular junction, and musculature. The risk of such adverse reactions is proportional to the concentration of LA achieved in the circulation. Plasma Concentration of Local Anesthetics The following factors determine the plasma concentration of LAs: Noted that although the peak level of a LA is directly related to the dose administered, administration of the same dose at different sites results in marked differences in peak blood levels. This explains why large doses of LA can be used with peripheral nerve blocks without the toxicity that would be seen with an intramuscular or intravenous (IV) injection. It is also for this reason that the adherence to strictly defined maximum doses of LAs is nonsensical. As an example, 150 mg of ropivacaine at the popliteal fossa will result in a markedly different plasma level than the same dose administered intercostally. Careful consideration of patient factors and the requirements of the block should precede selection of LA type and dose for peripheral nerve block. Short-acting ester local anesthetics are inherently safer with respect to systemic toxicity due to their clearance by pseudocholinesterase. In the case of 2-chloroprocaine, peak blood levels achieved are affected by the rate at which the LA drug undergoes biotransformation and elimination (plasma half-life of about 45 seconds-1 minute). In contrast, peak blood levels of amidelinked LA drugs are primarily the result of absorption. Central Nervous System Toxicity 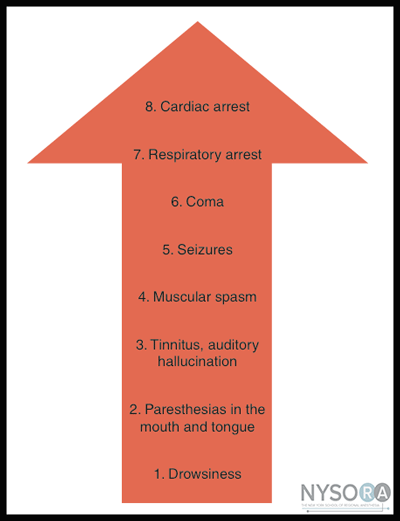 Figure 3: Progression of local anesthetic toxicity. The symptoms of CNS toxicity associated with LAs are a function of their plasma level (Figure 3). Toxicity is typically first expressed as stimulation of the CNS, producing restlessness, disorientation, and tremor. As the plasma concentration of the LA increases, tonic-clonic seizures occur; the more potent the LA, the more readily convulsions may be produced. With even higher levels of LA, central stimulation is followed by depression and respiratory failure, culminating in coma. The apparent stimulation and subsequent depression produced by applying LAs to the CNS presumably are due to depression of neuronal activity. Selective depression of inhibitory neurons is thought to account for the excitatory phase in vivo. However, rapid systemic administration of a LA may produce death with no, or only transient, signs of CNS stimulation. Under these conditions, the concentration of the drug probably rises so rapidly that all neurons are depressed simultaneously. Airway control and support of respiration constitute essential treatment. Benzodiazepines or a small dose of propofol (e.g., 0.5-1.0 mg/kg) administered IV in small doses are the drugs of choice for aborting convulsions. The use of benzodiazepines as premedication is often recommended to elevate the seizure threshold; however, they must be used with caution because respiratory depression with excessive sedation may produce respiratory acidosis with a consequent higher level of free drug in the serum. Cardiovascular Toxicity The primary site of action of LAs in the cardiovascular system is the myocardium, where they decrease electrical excitability, conduction rate, and the force of myocardial contraction. Most LAs also cause arteriolar dilatation, contributing to hypotension. Cardiovascular effects typically occur at higher systemic concentrations than those at which effects on the CNS are produced. However, note that it is possible for cardiovascular collapse and death to occur even in an absence of the warning signs and symptoms of CNS toxicity. It is believed that this is probably the result of action on the pacemaker cells or the sudden onset of ventricular fibrillation. In animal studies on LA cardiotoxicity, all caused dose-dependent depression of the contractility of cardiac muscle. This depressant effect on cardiac contractility parallels the anesthetic potency of the LA in blocking nerves. Therefore, bupivacaine, which is four times more potent than lidocaine in blocking nerves, is also four times more potent in depressing cardiac contractility. Deaths caused by a bupivacaine overdose have been associated with progressive prolongation of ventricular conduction and widening of the QRS complex, followed by the sudden onset of arrhythmia such as ventricular fibrillation. Pregnancy and Local Anesthetic Toxicity Plasma concentrations of AAG are also decreased in pregnant women and in newborns. This lowered concentration effectively increases the free fraction of bupivacaine in plasma, and it may have been an important contributing factor to the bupivacaine toxicity in pregnant patients and to the number of cardiac arrests that have been reported with inadvertent overdoses of bupivacaine in pregnant women. However, with intermediate-duration LAs (e.g., lidocaine and mepivacaine), smaller changes in protein binding occur during pregnancy, and the use of these LAs is not associated with an increased risk of cardiac toxicity during pregnancy. Pharmacodynamics and Treatment of Local Anesthetic Toxicity Blood levels of lidocaine associated with the onset of seizures appear to be in the range of 10 to 12 µg/mL. At these concentrations, inhibitory pathways in the brain are selectively disabled, and excitatory neurons can function unopposed. As the blood levels of lidocaine are increased further, respiratory depression becomes significant and at much higher levels (20-25 µg/mL), cardiotoxicity is manifested. In contrast, for bupivacaine, blood levels of approximately 4 µg/mL result in seizures, and blood levels between 4 and 6 µg/mL are associated with cardiac toxicity. This is reflective of a much lower therapeutic index for bupivacaine compared with lidocaine in terms of cardiac toxicity. In the setting of neurotoxicity without cardiac effects, high levels of LA in the brain rapidly dissipate and are redistributed to other tissue compartments. However, with the onset of significant cardiotoxicity, cardiac output diminishes, resulting in impairment in redistribution.
The long-acting LAs, such as bupivacaine and etidocaine, carry a significantly higher risk of cardiac arrest and difficult resuscitation. Note that such complications do not always occur immediately on injection of a LA and may be delayed up to 30 minutes. Many of these toxic effects occur following inadvertent intravascular injection of large amounts of drugs via an epidural catheter or from tourniquet failure during IV administration of a regional anesthetic. In patients with systemic neurotoxicity, treatment consists of halting the seizure by administering a benzodiazepine (midazolam 0.05-0.1 mg/kg) or a small dose of propofol (0.5-1.0 mg/kg) while preventing the detrimental effects of hypoxia and hypercarbia by ventilating with 100% oxygen. Providing that the hemodynamics are adequate, this can be achieved through bag-mask ventilation or the use of a laryngeal mask airway: Tracheal intubation is not always necessary (or always desirable) because these episodes are fortunately often short lived. However, it is extremely important to oxygenate and ventilate the patient because hypoxia, hypercarbia, and acidosis all potentiate the negative inotropic and chronotropic effects of LA toxicity. If ventilation is inadequate with the measures just described, tracheal intubation with the aid of a muscle relaxant is indicated. For patients exhibiting signs of cardiotoxicity (widened QRS complex, bradycardia, hypotension, ventricular fibrillation, and/or cardiovascular collapse), standard cardiac resuscitative measures should apply. Of primary importance in cardiac arrest is the maintenance of some degree of circulation and perfusion of vital organs by closed chest massage while electrical and pharmacologic therapies are being instituted. Drugs that have little value or indeed worsen the resuscitative efforts include bretylium, lidocaine, and calcium channel blockers. The use of lipid emulsion in LA toxicity has been reported to result in remarkably successful resuscitation, despite the fact that its mechanism remains obscure. Several theories exist, such as its role as a "lipid sink," that draws the drug out of solution, as well as its possible role in overriding the inhibition of mitochondrial carnitine-acylcarnitine translocase, thereby providing the myocardium with fatty acid for fuel. Although neither theory has been proven, the vast majority of experimental evidence suggests that lipid emulsion therapy is a low-risk, high-yield intervention that can only benefit the otherwise moribund patient (see Table 1 for directions for use). The use of vasopressor agents, such as epinephrine and vasopressin, has long been a standard part of the management of cardiac arrest, and at the moment, guidelines have not changed. However, experimental evidence involving animals has shown worsened outcomes when combined with lipid emulsion, perhaps due to increased myocardial intracellular acidosis. Further work in this area needs to be done before any recommendation can be made, and at present, the inclusion of these agents as standard resuscitative drugs is warranted. Finally, in cases of severe toxicity with large doses of long-acting LAs, timely institution of cardiopulmonary bypass may prove lifesaving. Types of Local Anesethetics As previously mentioned, LA drugs are classified as esters or amides (Figure 4). A short overview of various LAs with comments regarding their clinical applicability in peripheral blockade is provided in this section. Ester-Linked Local Anesthetics Ester-linked LAs are hydrolyzed at the ester linkage in plasma by pseudocholinesterase. The rate of hydrolysis of ester-linked LAs depends on the type and location of the substitution in the aromatic ring. For example, 2-chloroprocaine is hydrolyzed about four times faster than procaine, which in turn is hydrolyzed about four times faster than tetracaine. However, the rate of hydrolysis of all ester-linked LAs is markedly decreased in patients with atypical plasma pseudocholinesterase, and a prolonged epidural block in a patient with abnormal pseudocholinesterase has been reported. Another hallmark of metabolism of ester-linked LAs is that their hydrolysis leads to the formation of para-aminobenzoic acid (PABA). PABA and its derivatives carry a small risk potential for allergic reactions. A history of an allergic reaction to a LA should immediately suggest that a current reaction is due to the presence of PABA derived from an ester-linked LA. However, although exceedingly rare, allergic reactions can also develop from the use of multiple-dose vials of amide-linked LAs that contain PABA as a preservative. 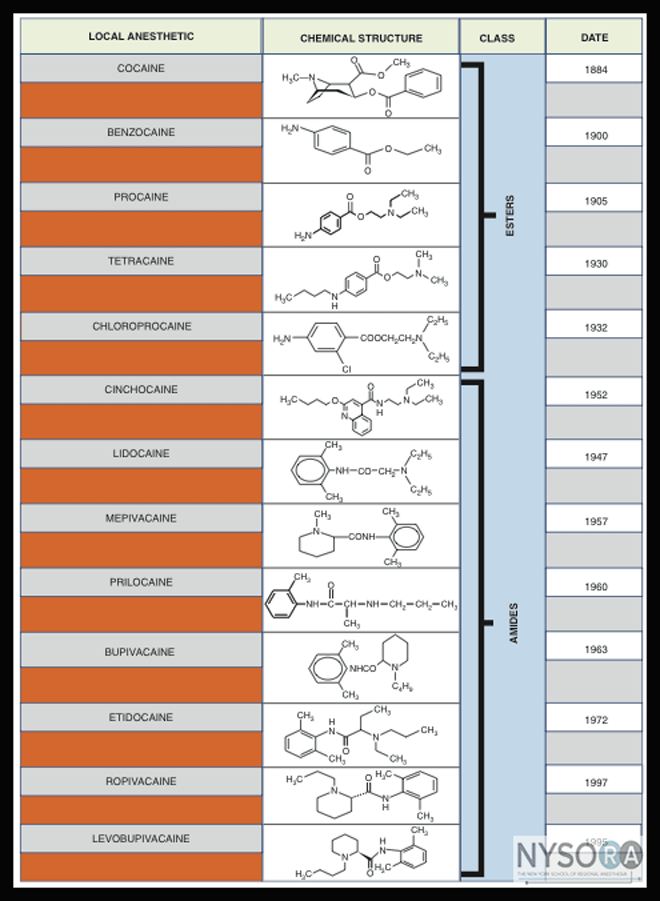
Figure 4: Local anesthetics, their classification, chemical structure, and approximate time of introduction. Cocaine Cocaine occurs naturally in the leaves of the coca shrub and is an ester of benzoic acid. The clinically desired actions of cocaine are blockade of nerve impulses and local vasoconstriction secondary to inhibition of local norepinephrine reuptake. However, its toxicity and the potential for abuse have precluded wider clinical use of cocaine in modern practice. Its euphoric properties are due primarily to inhibition of catecholamine uptake, particularly dopamine, at CNS synapses. Other LAs do not block the uptake of norepinephrine and do not produce the sensitization to catecholamines, vasoconstriction, or mydriasis characteristics of cocaine. Currently, cocaine is used primarily to provide topical anesthesia of the upper respiratory tract, where its combined vasoconstrictor and LA properties provide anesthesia and shrinking of the mucosa with a single agent. Procaine Procaine, an amino ester, was the first synthetic LA. Procaine is characterized by low potency, slow onset, and short duration of action. Consequently, although once widely used, its use now is largely confined to infiltration anesthesia and perhaps diagnostic nerve blocks. 2-Chloroprocaine An ester LA introduced in 1952, 2-chloroprocaine is a chlorinated derivative of procaine. Chloroprocaine is the most rapidly metabolized LA used currently. Because of its rapid breakdown in plasma ( Tetracaine Tetracaine was introduced in 1932, and it is a long-acting amino ester. It is significantly more potent and has a longer duration of action than procaine or 2-chloroprocaine. Tetracaine is more slowly metabolized than the other commonly used ester LAs, and it is considerably more toxic. Currently, it is used in spinal anesthesia when a drug of long duration is needed, as well as in various topical anesthetic preparations. Because of its slow onset and potential for toxicity, tetracaine is used rarely for peripheral nerve blocks in our practice. Some centers, however, have used tetracaine in combination with other local anesthetics, commonly lidocaine, with success. The combination is often known by the nickname "supercaine." The prevalence of the use of supercaine in modern regional anesthesia is not known. Amide-Linked Local Anesthetics As opposed to ester-linked drugs, amide-linked LAs are metabolized in the liver by a dealkalization reaction in which an ethyl group is cleaved from the tertiary amine. The hepatic blood flow and liver function determine the hepatic clearance of these anesthetics. Consequently, factors that decrease hepatic blood flow or hepatic drug extraction both result in an increased elimination half-life. Renal clearance of unchanged LAs is a minor route of elimination, accounting for only 3% to 5% of the total drug administered. Lidocaine Lidocaine was introduced in 1948 by the Swedish drug manufacturer Astra, and it remains one of the most versatile and widely used LAs. It is the prototype of the amide class of LAs. Lidocaine is absorbed rapidly after parenteral administration and from the gastrointestinal and respiratory tracts. Lidocaine can be used in almost any peripheral nerve block in which a LA of intermediate duration is needed. A concentration of 1.5% or 2% with or without the addition of epinephrine is most commonly used for surgical anesthesia. More diluted concentrations are suitable in pain management, particularly for diagnostic blocks. Mepivacaine Mepivacaine, introduced in 1957, is an intermediate-duration amino amide LA. Its pharmacologic properties are similar to those of lidocaine. Although it was suggested that mepivacaine is more toxic to neonates (and as such is not used in obstetric anesthesia), it appears to have a slightly higher therapeutic index in adults than lidocaine. Its onset of action is similar to that of lidocaine, but it enjoys a slightly longer duration of action than lidocaine. Our first choice in any peripheral nerve block technique is 1.5% mepivacaine when an intermediate-duration blockade is desired (3-6 hours, depending on the type of nerve block and addition of a vasoconstrictor). Prilocaine Prilocaine is an intermediate-duration amino amide LA with a pharmacologic profile similar to that of lidocaine. The primary differences are a lack of vasodilatation and an increased volume of distribution, which reduces its CNS toxicity. However, it is unique among amide LAs for its propensity to cause methemoglobinemia, an effect of metabolism of the aromatic ring to o-toluidine. The development of methemoglobinemia depends on the total dose administered (usually requires 8 mg/kg) and does not have significant consequences in healthy patients. If necessary, it is treated by IV administration of methylene blue (1-2 mg/kg). Prilocaine is used infrequently in peripheral nerve blockade. Etidocaine Etidocaine is a long-acting amino amide introduced in 1972. Its neuronal blocking properties are characterized by an onset of action similar to that of lidocaine and a duration of action comparable with that of bupivacaine. Etidocaine is structurally similar to lidocaine, with alkyl substitution on the aliphatic connecting group between the hydrophilic amine and the amide linkage. This feature increases the lipid solubility of the drug and results in a drug more potent than lidocaine that has a very rapid onset of action and a prolonged duration of anesthesia. A major disadvantage of etidocaine is its profound motor blockade over a wide range of clinical concentrations, which often outlasts sensory blockade. For these reasons, etidocaine is not used for peripheral nerve blockade. Bupivacaine Since its introduction in 1963, bupivacaine has been one of the most commonly used LAs in regional and infiltration anesthesia. Its structure is similar to that of lidocaine, except that the amine-containing group is a butylpiperidine. Bupivacaine is a long-acting agent capable of producing prolonged anesthesia and analgesia that can be prolonged even further by the addition of epinephrine. It is substantially more cardiotoxic than lidocaine. The cardiotoxicity of bupivacaine is cumulative and substantially greater than would be predicted by its LA potency. At least part of the cardiotoxicity of bupivacaine may be mediated centrally because direct injection of small quantities of bupivacaine into the medulla can produce malignant ventricular arrhythmias. Bupivacaine-induced cardiotoxicity can be difficult to treat. Bupivacaine is widely used both in neuraxial and peripheral nerve blockade. The blocking property is characterized by a slower onset and a long, somewhat unpredictable duration of blockade. Because of its toxicity profile, large doses of bupivacaine should be avoided. Ropivacaine The cardiotoxicity of bupivacaine stimulated interest in developing a less toxic, long-lasting LA. The development of ropivacaine, the S-enantiomer of 1-propyl-2', 6'-pipecolocylidide, is the result of that search. The S-enantiomer, like most LAs with a chiral center, was chosen because it has a lower toxicity than the R-enantiomer. This is presumably because of slower uptake, resulting in lower blood levels for a given dose. Ropivacaine undergoes extensive hepatic metabolism after IV administration, with only 1% of the drug eliminated unchanged in the urine. Ropivacaine is slightly less potent than bupivacaine in producing anesthesia when used in lower concentrations. However, in concentrations of 0.5% and higher, it produces dense blockade with a slightly shorter duration than that of bupivacaine. In concentrations of 0.75%, the onset of blockade is almost as fast as that of 1.5% mepivacaine or 3% 2-chloroprocaine, with reduced CNS toxicity and cardiotoxic potential and a lower propensity for motor blockade than bupivacaine. For these reasons, ropi- vacaine has become one of the most commonly used long acting LAs in peripheral nerve blockade.
Levobupivacaine Levobupivacaine contains a single enantiomer of bupivacaine hydrochloride, and is less cardiotoxic than bupivacaine. It is extensively metabolized with no unchanged drug detected in the urine or feces. The properties of levobupivacaine in peripheral nerve blockade are less well studied than those of ropivacaine, however, research results suggest that they seem to parallel those of bupivacaine. Therefore, levobupivacaine is a suitable, less toxic alternative to bupivacaine. Additives to Local Anesthetics Vasoconstrictors The addition of a vasoconstrictor to a LA delays its vascular absorption, increasing the duration of drug contact with nerve tissues. The net effect is prolongation of the blockade by as much as 50% and a decrease in the systemic absorption of LA. These effects vary significantly among different types of LAs and individual nerve blocks. For example, because lidocaine is a natural vasodilator, the effect is pronounced for those blocks compared with blocks using ropivacaine, which has its own slight vasoconstricting effect. Epinephrine is the most commonly used vasoconstrictor in peripheral nerve blockade. A decrease in nerve blood supply has been associated with epinephrine when combined with local anesthetics. However, this effect was not seen when concentrations of epinephrine were maintained at 1:400,000 (2.5 µg/mL). As such, this is the recommended concentration when used as an adjuvant.
Opioids The injection of opioids into the epidural or subarachnoid space to manage acute or chronic pain is based on the knowledge that opioid receptors are present in the substantia gelatinosa of the spinal cord. Thus combinations of a LA and an opiate are often successfully used in neuraxial blockade to both enhance the blockade and prolong analgesia. However, in peripheral nerves, similar receptors are absent or the effects of opiates are negligibly weak. For this reason, opiates do not have a significant clinical role in peripheral nerve blockade. Clonidine Clonidine is a centrally acting selective partial alpha-2-adrenergic agonist. Because of its ability to reduce sympathetic nervous system output from the CNS, clonidine acts as an antihypertensive drug. Preservative-free clonidine, administered into the epidural or subarachnoid space (150-450 µg), produces dose-dependent analgesia and, unlike opioids, does not produce depression of ventilation, pruritus, nausea, or vomiting. Clonidine produces analgesia by activating postsynaptic α2 receptors in the substantia gelatinosa of the spinal cord. Much less research has been done on the effects of clonidine in peripheral nerve blockade. A recent meta-analysis showed that clonidine prolongs the duration of both the sensory and motor block by approximately 1.5 to 2 hours. Of note, there appears to be no benefit to using clonidine in continuous perineural infusions. In addition, side effects, notably sedation, orthostatic hypotension, and fainting, should be considered when using clonidine. The latter two effects, in particular, can interfere with mobilization postoperatively. Although life-threatening hypotension or bradycardia has not been reported when clonidine is used with peripheral nerve blocks, its circulatory effects may complicate resuscitation in a setting of LA toxicity. Selecting Local Anesthetics for Peripheral Nerve Blocks With a variety of LAs to choose from, it is useful to keep the following points in mind when selecting an agent for nerve blockade. In general, the onset and duration of a LA are similar in nature. For example, 2-chloroprocaine has a short onset but also a relatively brief duration (Table 1). In contrast, bupivacaine and ropivacaine have longer durations of action but take somewhat longer to exert their effect. The LA should be tailored to the duration of the surgical procedure and the anticipated degree of pain. For example, creation of an arteriovenous fistula is a relatively short operation with minimal postoperative pain. Therefore, selection of a short-acting agent (e.g., mepivacaine) provides excellent intraoperative conditions, without the burden of an insensate limb for 10 to 18 hours postoperatively. A rotator cuff repair involves a greater degree of postoperative pain, and therefore selection of a long-acting LA (e.g., ropivacaine) is appropriate. Onset and duration for a given LA varies according to the nerve or plexus blocked. For example, at the brachial plexus, 0.5% ropivacaine may be expected to provide 10 to 12 hours of analgesia; the same concentration at the sciatic nerve may provide up to 24 hours of analgesia. This is likely due to differences in the local vascularity, which influences the uptake of LA. Blocks for postoperative analgesia (often in concert with general anesthesia) do not require a high concentration of LA. Ropivacaine 0.2% is usually sufficient to provide excellent sensory analgesia but spare any motor blockade. The toxicity of the agent should be considered. Bupivacaine provides the longest duration of the commonly used LAs but also has the worst cardiotoxic profile. However, a reduction in the dose used, seen more and more frequently with ultrasound-guided nerve blocks, may provide an equivalent or greater safety profile than a larger dose of ropivacaine, the drug to which it is often compared. Table 1: Choice of Local Anesthetic for Peripheral Nerve Blockade  Ambulatory patients who are undergoing lower limb surgery should be administered long-acting LAs with caution and with proper education regarding ambulation with assistance. Short procedures with minimal postoperative pain (most ambulatory procedures) only require a short- to intermediate-acting agent, and evidence of block resolution is often a requirement before discharge home. Anticipated pain lasting longer than 15 to 20 hours should warrant the consideration of a perineural catheter. The appreciation that patients show for any effective block will fade quickly when it wears off in the middle of the night, leaving them unprepared, in pain, and unable to access alternative pain therapies. Mixing of Local Anesthetics Mixing of LAs (e.g., lidocaine and bupivacaine) is often done in clinical practice with the intent of obtaining the faster onset of the shorter acting LAs and the longer duration of the longer acting LA. Unfortunately, when LAs are mixed, their onset, duration, and potency become much less predictable, and the end result is far from expected. For instance, work performed at our institution has shown that combining mepivacaine 1.5% with bupivacaine 0.5%, versus either drug alone for interscalene brachial plexus block, results in little clinical advantage. Onset times for all three solutions were indistinguishable, and the duration of the combined solution was significantly shorter than bupivacaine alone. Moreover, a separate experiment looking at the sequence of LAs (i.e., bupivacaine first, mepivacaine second versus mepivacaine first, bupivacaine second) showed no difference in the onset and duration between groups. Therefore, if long duration is desired, a long-acting drug alone will provide the best conditions. In addition, mixing LAs also carries a risk of a drug error. For this reason, we rarely mix LAs; instead, we choose drugs and concentrations of single agents to achieve the desired effects. The vast majority of nerve block goals can be met using one short/intermediate and one long-acting LA. At our institution, more than 90% of peripheral nerve blocks for surgical anesthesia are performed with one of two LAs: 1.5% mepivacaine or 0.5%-0.75% ropivacaine. Of course, there are times when other LAs are used. Chloroprocaine is used for quick knee arthroscopies, for example, when the postoperative pain is minimal and rapid return to ambulation is required. Table 1 shows the commonly used local anesthetics, with and without bicarbonate and/or epinephrine, and their expected onset and duration of actions. As mentioned previously, these numbers do not apply to all scenarios, all nerves, and all plexuses but can be used as a rough comparative guide to aid in decision making.
|


 Facebook
Facebook
 del.icio.us
del.icio.us
 Digg
Digg
 StumbleUpon
StumbleUpon
More from Foundations of RA
Equipment for Peripheral Nerve Blocks
Ali Nima Shariat, Patrick M. Horan, Kimberly Gratenstein, Colleen McCally, and Ashton P. Frulla Introduction ...

Indications for Peripheral Nerve Blocks
AUTHOR: Jeff Gadsden Introduction During the past 20 years, increasing knowledge in functional regional anesthesia anatomy, coupled ...

Continuous Peripheral Nerve Blocks in Outpatients
Introduction - Single-injection nerve blocks provide up to 16 hours of postoperative analgesia - Portable infusion pumps ...
Neurologic Complications of Peripheral Nerve Blocks
Author: Jeff Gadsden Nerve injury following peripheral nerve blockade (PNB) is a potentially devastating complication that ...

Toxicity of Local Anesthetics
AUTHORS: Steven Dewaele, Alan C. Santos Introduction Systemic toxicity of local anesthetics can occur after administration of ...
Essentials of Regional Anesthesia Anatomy
--- Authors: Admir Hadzic and Carlo Franco A good practical knowledge of anatomy is important ...
Monitoring and Documentation
AUTHOR: Jeff Gadsden Introduction The incidence of complications from general anesthesia has diminished substantially in recent ...
Electrical Nerve Stimulators and Localization of Peripheral Nerves
Ali Nima Shariat, Patrick M. Horan, Kimberly Gratenstein, Colleen McCally, and Ashton P. Frulla History of ...
| 12/19/2015(+ 2016 Dates) | |
| 01/27/2016 | |
| 03/17/2016 | |
| 04/20/2016 | |
| 09/24/2016 | |
| 10/01/2024 |
![[advertisement] gehealthcare](../../files/banners/banner1_250x600/GEtouch(250X600).gif)
Image gallery







Post your comment